








精密定量プロテオミクスを用いたシグナル伝達の包括的解析
 研究代表者
研究代表者
松本雅記
九州大学 生体防御医学研究所 プロテオミクス分野
http://www.bioreg.kyushu-u.ac.jp/mib/divisions/summary_proteomics.pdf
研究概要
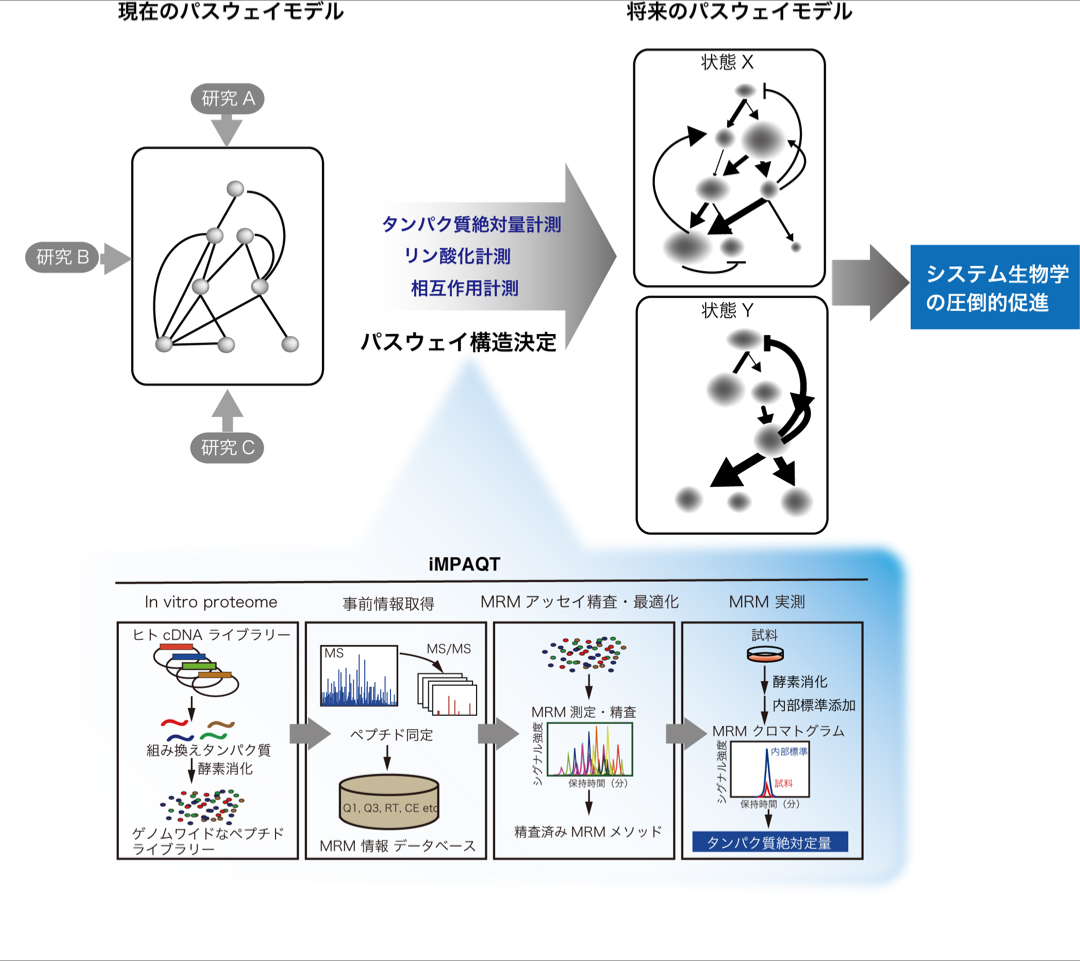
細胞運命決定・増殖などの本質的な生命現象は細胞外刺激が引き起こす細胞内シグナル伝達によって規定されています。これまでの研究からシグナル伝達に関与するタンパク質が多数同定され、それらの間で起きる相互作用やリン酸化反応を介してシグナル伝達経路が形成されていることがわかっています。一方で、同一シグナル経路が刺激の種類や強さによって異なる出力(増殖や分化などの細胞応答)を生み出す機構などはほとんど分かっていません。このようにシグナル伝達の特性や動作原理に関するシステム論的解析はまさに始まったばかりの新しい研究領域です。近年、シグナル伝達を数理モデルに落とし込んで理解する試みがなされていますが、シグナル伝達因子の濃度情報やリン酸化をはじめとする飜訳後修飾の変化に関する定量的な情報が不足していることが大きな問題となっています。本研究ではこのようなシグナル伝達の数理モデリングにおける重要課題を解決すべく、独自に開発してきた革新的なタンパク質絶対定量法iMPAQT (in vitro proteome assisted MRM for protein absolute quantification) 法をパスウェイ構造決定へ応用するものです。iMPAQT法は網羅的に合成した組み換えタンパク質を対象にLC-MS/MS解析を事前に実施し、得られた各ペプチドの座標(LC上の保持時間、質量、部分質量)のデータベース化を行なっています。したがって、これらの情報を元にしてMRM (multiple reaction monitoring) 法を容易に実施可能です。iMPAQT法は従来法に比べて圧倒的な感度を有し(検出限界:103〜104分子/1細胞)、シグナル伝達分子などの低発現タンパク質も一網打尽に検出可能です。つまり、多数のタンパク質で構成される細胞内シグナル伝達ネットワークを定量的に掌握できる優れた手法であると言えます。本手法はタンパク質の発現絶対量だけでなく、細胞内タンパク質ネットワーク動態(リン酸化などの翻訳後修飾や相互作用の変化)の解析にも応用可能です。本研究ではiMPAQT法を駆使してシグナル伝達の包括的解析を行い、シグナル伝達の特異性や多様性を生み出す動作原理の解明を目指します。
参考文献
- Yachie N, Robotic Biology Consortium (Inc. Masaki Matsumoto), Natsume T. Robotic Crowd Biology: LabDroids accelerate life science experiments. Nature Biotech. 35, 310-312 (2017).
- Matsumoto M, Matsuzaki F, Oshikawa K, Goshima N, Mori M, Kawamura Y, Ogawa K, Fukuda E, Nakatsumi H, Natsume T, Fukui K, Horimoto H, Nagashima T, Funayama R, Nakayama K, Nakayama KI. Development of large-scale targeted proteomics assay resource based on an in vitro human proteome. Nature Methods 14, 251-258 (2017).
- Matsumoto A, Pasut A, Matsumoto M, Yamashita R, Fung J, Monteleone E, Saghatelian A, Nakayama KI, Clohessy JG, Pandolfi PP. mTORC1 and muscle regeneration are regulated by the LINC00961-encoded SPAR polypeptide. Nature. 541, 228-232 (2017).
- Okuda S, Watanabe Y, Moriya Y, Kawano S, Yamamoto T, Matsumoto M, Takami T, Kobayashi D, Araki N, Yoshizawa AC, Tabata T, Sugiyama N, Goto S, Ishihama Y. jPOSTrepo: an international standard data repository for proteomes. Nucleic Acids Res. D45, D1107-D1111 (2017).
- Hatano A, Matsumoto M, Nakayama, KI. Phosphoproteomics analyses show subnetwork systems in T-cell receptor signaling. Genes Cells 21, 1095-1112 (2016).
- Yugi K, Kubota H, Toyoshima Y, Noguchi R, Kawata K, Komori Y, Uda S, Kunida K, Tomizawa Y, Funato Y, Miki H, Matsumoto M, Nakayama KI, Kashikura K, Endo K, Ikeda K, Soga T, Kuroda S. Reconstruction of insulin signal flow from phosphoproteome and metabolome data. Cell Rep. 8, 1171-1183 (2014).
- Hirano A., Yumimoto K, Tsunematsu R, Matsumoto M, Oyama M, Kozuka-Hata H, Nakagawa T, Lanjakornsiripan D, Nakayama KI, Fukada Y. FBXL21 regulates oscillation of the circadian clock through ubiquitination and stabilization of cryptochromes. Cell 152, 1106-1118 (2013).
- Narumi R, Murakami T, Kuga T, Adachi J, Shiromizu T, Muraoka S, Kume H, Kodera Y, Matsumoto M, Nakayama K, Miyamoto Y, Ishitobi M, Inaji H, Kato K, Tomonaga T. A strategy for large-scale phosphoproteomics and SRM-based validation of human breast cancer tissue samples. J. Proteome Res. 11, 5311-5322 (2012).
- Yumimoto K, Matsumoto M, Oyamada K, Moroishi T, Nakayama KI. Comprehensive identification of substrates for F-box proteins by differential proteomics analysis. J. Proteome Res. 11, 3175-3185 (2012).
- Oshikawa K, Matsumoto M, Oyamada K, Nakayama KI. Proteome-wide identification of ubiquitylation sites by conjugation of engineered lysine-less ubiquitin. J. Proteome Res. 11, 796-807 (2012).
![]()